
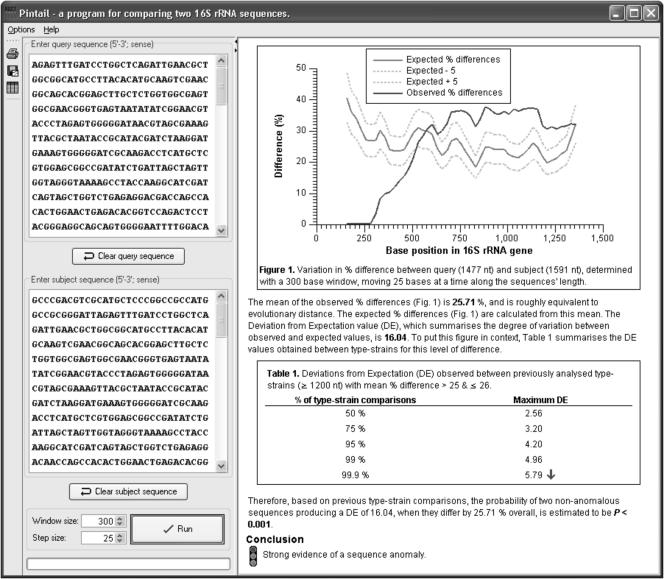
A brand new methodology for detecting chimeras and different anomalies inside 16S rRNA sequence records is offered.
Using this methodology, we screened 1,399 sequences from 19 phyla, as outlined by the Ribosomal Database Project, launch 9, replace 22, and located 5.0% to harbor substantial errors. Of these, 64.3% had been apparent chimeras, 14.3% had been unidentified sequencing errors, and 21.4% had been extremely degenerate.
In all, 11 phyla contained apparent chimeras, accounting for 0.8 to 11% of the records for these phyla. Many chimeras (43.1%) had been shaped from parental sequences belonging to completely different phyla.
While most comprised two fragments, 13.7% had been composed of at least three fragments, usually from three completely different sources. A separate evaluation of the Bacteroidetes phylum (2,739 sequences) additionally revealed 5.8% records to be anomalous, of which 65.4% had been apparently chimeric. Overall, we conclude that, as a conservative estimate, 1 in each 20 public database records is possible to be corrupt.
Our outcomes help considerations lately expressed over the standard of the public repositories.
With 16S rRNA sequence information more and more enjoying a dominant function in bacterial systematics and environmental biodiversity research, it is very important that steps be taken to enhance screening of sequences prior to submission.

To this finish, we’ve got applied our methodology as a program with a simple-to-use graphic person interface that is able to operating on a variety of laptop platforms.
The program is known as Pintail, is launched beneath the phrases of the GNU General Public License open supply license, and is freely accessible from our web site at http://www.cardiff.ac.uk/biosi/research/biosoft/.
Multiple endocrine neoplasia sort 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations.
Multiple endocrine neoplasia sort 2 (MEN2) is an inherited, autosomal-dominant dysfunction brought on by deleterious mutations inside the RET protooncogene. MEN2 RET mutations are primarily heterozygous, missense sequence adjustments discovered in RET exons 10, 11, and 13-16.
Our group has developed the publicly accessible, searchable MEN2 RET database to help in genotype/phenotype correlations, utilizing Human Genome Variation Society suggestions for sequence variation nomenclature and database content material. The MEN2 RET database catalogs all RET sequence variation related to the MEN2 syndromes, with related medical data.
Each database entry lists a RET sequence variation’s location inside the RET gene, genotype, pathogenicity classification, MEN2 phenotype, first literature reference, and feedback (which can contain data on different medical options, advanced genotypes, and extra literature references).
e MEN2 phenotype definitions had been derived from the International RET Mutation Consortium pointers for classification of MEN2 illness phenotypes. Although practically the entire 132 RET sequence variation entries initially cataloged in the database had been from literature experiences, novel sequence variation and up to date phenotypic data for any current database entry will be submitted electronically on the database web site.
The database web site additionally accommodates hyperlinks to chosen MEN2 literature critiques, gene and protein data, and RET reference sequences. The MEN2 RET database (www.arup.utah.edu/database/MEN2/MEN2_welcome.php) will function a repository for MEN2-associated RET sequence variation and reference for RET genotype/MEN2 phenotype correlations.